
Биотехнологии →
Практическая биоинформатика ч. 2
Эта статья расскажет о том, как обработать данные, полученные после pipeline, выходом которого будет sam/bam файл[1], создать несложный bed graph файл (http://genome.ucsc.edu/FAQ/FAQformat.html) и просмотреть его с помощью UCSC genome browser[2]. Очень сложно решиться, на чем писать программы, ибо уже есть огромное количество чужих наработок и совсем не хочется сочинять колесо там, где этот этап уже пройден. Долго мучаясь, я решил остановиться на C++, хотя phyton и R рассматривались на равных. Также сохранилась идея, что может понадобиться графика, да ещё и под Linux, поэтому к С++ прибавилось Qt. Надеюсь, в этой статье я расскажу достаточно подробно о всем выше перечисленном, чтобы ответить на вопрос, заданный мне в начале пути и озвученный в первой части повествования.
Для работы с sam/bam файлами нам понадобится собранный пакет samtools[1]. Скачиваем пакет с сайта samtools.sourceforge.net/, разворачиваем в директорию, например, /usr/src заходим в создавшуюся директорию и набираем make. У меня в системе не было установлено XCurses и я заменил строчку “LIBCURSES= -lXCurses” на “LIBCURSES= -lncurses” и все собралось. Результатом работы программы make стала собранная программа samtools и библиотека libbam.a.
Нам нужны C++ классы, которые будут хранить информацию о ридах, генах, интронах, эксонах и т.д. Для организации таких классов я воспользовался boost.intervals, хотя и не совсем в том виде, как хотелось. Ни boost.intervals, ни boost.icl не позволяют сохранять полную информацию об отрезках. Мне нужна следующая информация о множестве отрезков:
В частности, эта информация ответит на вопрос, какова высота покрытия отрезками текущего участка координатной оси.
С каждой новой статьей я буду пополнять и изменять эти классы. Сейчас их достаточно, чтобы собрать программу и прикинуть чего не хватает для будующего. Вот пример получившихся классов.
Reads.hpp
Теперь приступим к обертке над функциями для работы с sam файлами. Отличие между sam и bam заключается в том, что первый — это обычный текстовый файл, хоть и форматированный, а второй — сжатый и бинарный, т.е. структурированный. Я предпочитаю работать с bam при большом количестве ридов, sam файл может достигать гигабайтов.
BEDHandler.hpp
BEDHandler.cpp
Оставшуюся часть кода я приводить в статье не буду, вот ссылка на архив, структура довольно простая: в корне две директории thirdparty и src, в первой лежит samtools, во второй — sam2bedgraph и global. Для того, чтобы собрать проект, надо в директории sam2bedgraph запустить qmake и затем make. Проверял под openSUSE 12.1 x64 с родной Qt (4.7.4) и boost (1.46.1).
Ссылка на описание структуры bedgraph файла приведена в первом абзаце, но вкратце упомяну. Первая строчка задает характеристики файла, если присутствует. Тело файла содержит как минимум 4 столбца. В первом столбце указывается название хромосомы, во втором и третьем — координаты начала и конца отрезка («окна»), соответственно. В последнем столбце — «высота», в нашем случае это количество начал ридов на это окно. Размеры «окна» для каждого файла лучше подбирать экспериментально. Я для ChIP-seq использую окно в 200 bp, для RNA-seq окно в 20 bp.
У программы может быть два параметра: входной .bam файл и выходной файл, куда запишутся данные о bedgraph. Параметры можно и не задавать, тогда программа попытается открыть файл по умолчанию input.bam и создать выходной output.data. В домашней директории пользователя будет создана директория с конфигурационным файлом .config/CCHMC/sam2bedgraph.ini, в котором можно поменять значения имен файлов по умолчанию, изменить длину «окна», для которого считаем высоты и задать вид первой информационной строчки.
Получившийся файл можно загрузить в genome browser. Идем на сайт genome.ucsc.edu/[2] нажимаем на ссылку Genomes.
Попадаем на следующую страницу, где необходимо задать условия, при которых был получен наш bam файл: Clade(Класс), genome (Геноме), assembly (Аннотация, Сборка). И затем нажать кнопку “add custom tracks”.
Теперь можно нажать кнопку «Choose file» и загрузить наш файл, нажав кнопку «Submit». В результате попадаем на страницу с аннотацией, на которую будет выведена информация из базы данных и из нашего bedgraph. Если установить genome browser локально, то можно добавлять bedgraph на сайт не временно в рамках сессии, а постоянно. Можно организовывать специальные линки на сайт, в которых будет указано, где брать bedgraph файл. На сайте приведена инструкция, как скопировать genome browser.
Можно загрузить несколько файлов, что я и сделал. Результат вы видите на следующем скриншоте.
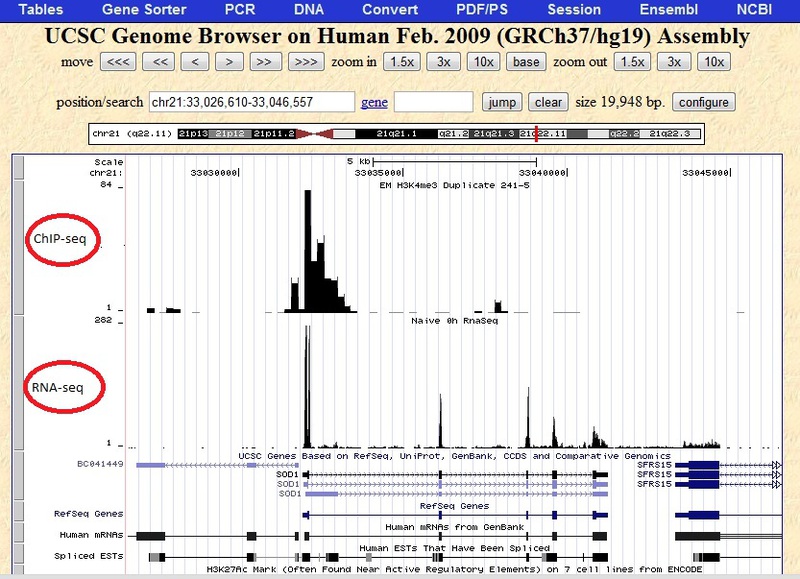
Как видно на этом скриншоте, есть участки с нулевым уровнем, а есть участки с обогащением, там где пики. Иногда виден фон или шум. Также наглядно видно, чем отличается ChIP-seq от RNA-seq. Определение этих участков из программы — отдельный вопрос, однозначного ответа на который нет. Дело в том, что пиков может быть столько, сколько генов в геноме или, и того хуже, сколько экзонов. И отделить обогащённые участки в рамках эксперимента очень трудоемко.
1. Li, H., et al., The Sequence Alignment/Map format and SAMtools. Bioinformatics, 2009. 25(16): p. 2078-9.
2. Kent, W.J., et al., The human genome browser at UCSC. Genome Res, 2002. 12(6): p. 996-1006.
3. Dreszer, T.R., et al., The UCSC Genome Browser database: extensions and updates 2011. Nucleic Acids Res, 2012. 40(Database issue): p. D918-23.
Для работы с sam/bam файлами нам понадобится собранный пакет samtools[1]. Скачиваем пакет с сайта samtools.sourceforge.net/, разворачиваем в директорию, например, /usr/src заходим в создавшуюся директорию и набираем make. У меня в системе не было установлено XCurses и я заменил строчку “LIBCURSES= -lXCurses” на “LIBCURSES= -lncurses” и все собралось. Результатом работы программы make стала собранная программа samtools и библиотека libbam.a.
Нам нужны C++ классы, которые будут хранить информацию о ридах, генах, интронах, эксонах и т.д. Для организации таких классов я воспользовался boost.intervals, хотя и не совсем в том виде, как хотелось. Ни boost.intervals, ни boost.icl не позволяют сохранять полную информацию об отрезках. Мне нужна следующая информация о множестве отрезков:
- любая пара начало/конец, начало/длина;
- все отрезки закрытые, т.е. включают концы;
- сколько отрезков начинается в заданной точке;
- сколько отрезков пересекается в точке;
- сколько отрезков начинается в заданном отрезке или пересекается с заданным отрезком.
В частности, эта информация ответит на вопрос, какова высота покрытия отрезками текущего участка координатной оси.
С каждой новой статьей я буду пополнять и изменять эти классы. Сейчас их достаточно, чтобы собрать программу и прикинуть чего не хватает для будующего. Вот пример получившихся классов.
Reads.hpp
#ifndef _READS_29122011_HPP_
#define _READS_29122011_HPP_
#include <boost/numeric/interval.hpp>
#include <QString>
#include <QMap>
#include <QtDebug>
namespace genome {
namespace bni = boost::numeric;
typedef bni::interval<int> read_position;
/**********************************************************************************
**********************************************************************************/
class Read
{
public:
Read():
multiplying(1),
length(0){};
Read(Read const &r):
multiplying(r.multiplying),
length(r.length),
position(r.position){
sentenceRepresentation=r.sentenceRepresentation;
qualityRepresentation=r.qualityRepresentation;};
Read(int start,int len,QString sr="",QString qr=""):
multiplying(1),
length(len),
position(start,start+len-1),
sentenceRepresentation(sr),
qualityRepresentation(qr){};
int getLevel() {return multiplying;};
void plusLevel() {++multiplying;};
int getStart() {return position.lower();};
int getLength() {return length;};
void operator+= (const int& c) {this->multiplying+=c;};
bool operator== (const Read& r) const {return this->position==r.position;};
bool operator!= (const Read& r) const {return this->position!=r.position;};
void operator++ (int) {this->multiplying++;};
private:
int multiplying;
int length;
read_position position;
QString sentenceRepresentation;
QString qualityRepresentation;
};
/**********************************************************************************
**********************************************************************************/
typedef QMap<int,Read> cover_map;
class Cover
{
public:
Cover():max_len(0){};
void add(Read&);
int getHeight(int);
int getHeight(int,int);
int getStarts(int);
int getStarts(int,int);
QList<int> getStarts();
cover_map::iterator getBeginIterator(){return covering.begin();};
cover_map::iterator getEndIterator(){return covering.end();};
bool operator== (const Cover& c) const {return this==&c;};
bool isEmpty(){return covering.size()==0;};
// static Cover empty(){ return Cover();};
private:
cover_map covering;
int max_len;
};
/**********************************************************************************
**********************************************************************************/
class Lines
{
public:
Lines(){};
Lines(Lines&){};
void addLine(QString, Read&);
Cover& getLineCover(QString);
QList<QString> getLines(void)
{
return lines.keys();
};
/*
*/
private:
QMap<QString,Cover> lines;
};
/**********************************************************************************
**********************************************************************************/
class GenomeDescription:
public Lines
{
public:
quint64 notAligned; // number of reads (ussualy form sam/bam file) that are not aligned
quint64 total;
/*
*/
void setGene(const QChar &sense,const QString &chrName,const qint32 &pos,const qint32 &,const qint32 &len)
{
Read r(pos,len);
addLine(chrName+sense,r);
};
/*
*/
GenomeDescription():Lines(),
notAligned(0),
total(0)
{};
};
}
#endif
Теперь приступим к обертке над функциями для работы с sam файлами. Отличие между sam и bam заключается в том, что первый — это обычный текстовый файл, хоть и форматированный, а второй — сжатый и бинарный, т.е. структурированный. Я предпочитаю работать с bam при большом количестве ридов, sam файл может достигать гигабайтов.
BEDHandler.hpp
#ifndef BEDHandler_H
#define BEDHandler_H
#include <config.hpp>
#include <Reads.hpp>
template <class Storage, class Result>
class BEDHandler : public QState
{
private:
Storage *sam_input;
Result *output;
QSettings setup;
QFile _outFile;
public:
BEDHandler(Storage &sam,Result &output,QState *parent=0);
~BEDHandler();
protected:
virtual void onEntry(QEvent* event);
};
#include <BEDHandler.cpp>
#endif
BEDHandler.cpp
//-------------------------------------------------------------
//-------------------------------------------------------------
template <class Storage,class Result>
BEDHandler<Storage,Result>::~BEDHandler()
{
}
//-------------------------------------------------------------
//-------------------------------------------------------------
template <class Storage,class Result>
BEDHandler<Storage,Result>::BEDHandler(Storage& sam,Result &_output,QState * parent):
QState(parent),
sam_input(&sam),
output(&_output)
{
_outFile.setFileName(setup.value("outFileName").toString());
_outFile.open(QIODevice::WriteOnly|QIODevice::Truncate);
}
//-------------------------------------------------------------
//-------------------------------------------------------------
template <class Storage,class Result>
void BEDHandler<Storage,Result>::onEntry(QEvent*)
{
if(!setup.contains("graphWindow"))
setup.setValue("graphWindow",200);
if(!setup.contains("siteShift"))
setup.setValue("siteShift",75);
if(!setup.contains("separateStrand"))
setup.setValue("separateStrand",false);
if(!setup.contains("HeaderString"))
setup.setValue("HeaderString","track type=bedGraph name=%1");
if(setup.value("HeaderString").toString()!="")
{
_outFile.write((setup.value("HeaderString").toString().arg(_outFile.fileName())+"\n").toLocal8Bit());
_outFile.flush();
}
quint32 window=setup.value("graphWindow").toUInt();
quint32 shift= setup.value("siteShift").toUInt();
QString line;
QString chrome;
foreach(line,sam_input->getLines())
{
if(line.endsWith("-")) continue;
chrome=line;
chrome.truncate(line.length()-1);
QMap <int,int> bed;
{
genome::cover_map::iterator i=sam_input->getLineCover(chrome+QChar('+')).getBeginIterator();
genome::cover_map::iterator e=sam_input->getLineCover(chrome+QChar('+')).getEndIterator();
while(i!=e)
{
int val=i.key()+shift;
bed[val-val%window]+=i.value().getLevel();
++i;
}
}
{
genome::cover_map::iterator i=sam_input->getLineCover(chrome+QChar('-')).getBeginIterator();
genome::cover_map::iterator e=sam_input->getLineCover(chrome+QChar('-')).getEndIterator();
while(i!=e)
{
int val=i.key()-shift;
if(val<0) val=0;
bed[val-val%window]+=i.value().getLevel();
++i;
}
}
QMap<int,int>::iterator i = bed.begin();
for(;i!=bed.end();i++)
{
_outFile.write(QString(chrome+"\t%1\t%2\t%3\n").
arg(i.key()).arg(i.key()+window).arg(i.value()).toLocal8Bit());
_outFile.flush();
}
}
}//end of function
Оставшуюся часть кода я приводить в статье не буду, вот ссылка на архив, структура довольно простая: в корне две директории thirdparty и src, в первой лежит samtools, во второй — sam2bedgraph и global. Для того, чтобы собрать проект, надо в директории sam2bedgraph запустить qmake и затем make. Проверял под openSUSE 12.1 x64 с родной Qt (4.7.4) и boost (1.46.1).
Ссылка на описание структуры bedgraph файла приведена в первом абзаце, но вкратце упомяну. Первая строчка задает характеристики файла, если присутствует. Тело файла содержит как минимум 4 столбца. В первом столбце указывается название хромосомы, во втором и третьем — координаты начала и конца отрезка («окна»), соответственно. В последнем столбце — «высота», в нашем случае это количество начал ридов на это окно. Размеры «окна» для каждого файла лучше подбирать экспериментально. Я для ChIP-seq использую окно в 200 bp, для RNA-seq окно в 20 bp.
У программы может быть два параметра: входной .bam файл и выходной файл, куда запишутся данные о bedgraph. Параметры можно и не задавать, тогда программа попытается открыть файл по умолчанию input.bam и создать выходной output.data. В домашней директории пользователя будет создана директория с конфигурационным файлом .config/CCHMC/sam2bedgraph.ini, в котором можно поменять значения имен файлов по умолчанию, изменить длину «окна», для которого считаем высоты и задать вид первой информационной строчки.
Получившийся файл можно загрузить в genome browser. Идем на сайт genome.ucsc.edu/[2] нажимаем на ссылку Genomes.
Попадаем на следующую страницу, где необходимо задать условия, при которых был получен наш bam файл: Clade(Класс), genome (Геноме), assembly (Аннотация, Сборка). И затем нажать кнопку “add custom tracks”.
Теперь можно нажать кнопку «Choose file» и загрузить наш файл, нажав кнопку «Submit». В результате попадаем на страницу с аннотацией, на которую будет выведена информация из базы данных и из нашего bedgraph. Если установить genome browser локально, то можно добавлять bedgraph на сайт не временно в рамках сессии, а постоянно. Можно организовывать специальные линки на сайт, в которых будет указано, где брать bedgraph файл. На сайте приведена инструкция, как скопировать genome browser.
Можно загрузить несколько файлов, что я и сделал. Результат вы видите на следующем скриншоте.
Как видно на этом скриншоте, есть участки с нулевым уровнем, а есть участки с обогащением, там где пики. Иногда виден фон или шум. Также наглядно видно, чем отличается ChIP-seq от RNA-seq. Определение этих участков из программы — отдельный вопрос, однозначного ответа на который нет. Дело в том, что пиков может быть столько, сколько генов в геноме или, и того хуже, сколько экзонов. И отделить обогащённые участки в рамках эксперимента очень трудоемко.
1. Li, H., et al., The Sequence Alignment/Map format and SAMtools. Bioinformatics, 2009. 25(16): p. 2078-9.
2. Kent, W.J., et al., The human genome browser at UCSC. Genome Res, 2002. 12(6): p. 996-1006.
3. Dreszer, T.R., et al., The UCSC Genome Browser database: extensions and updates 2011. Nucleic Acids Res, 2012. 40(Database issue): p. D918-23.
28.01.2012 00:43+0400